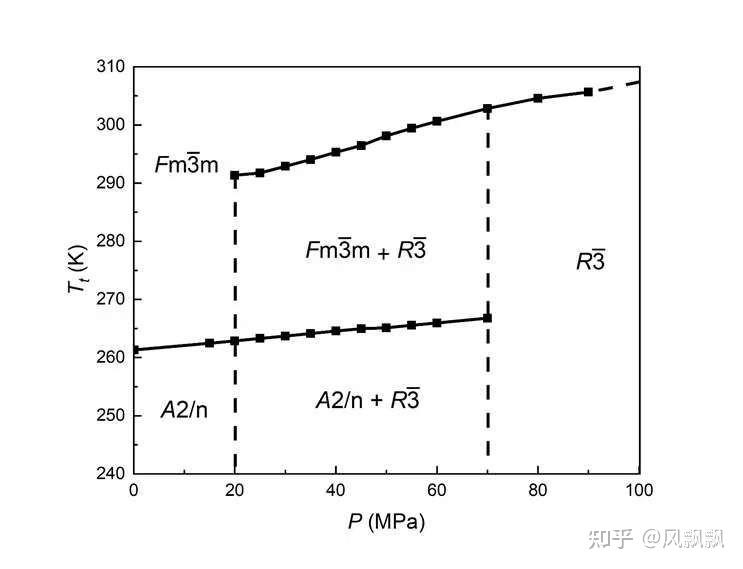
Phase diagram of KPF6 (potassium hexafluorophosphate)
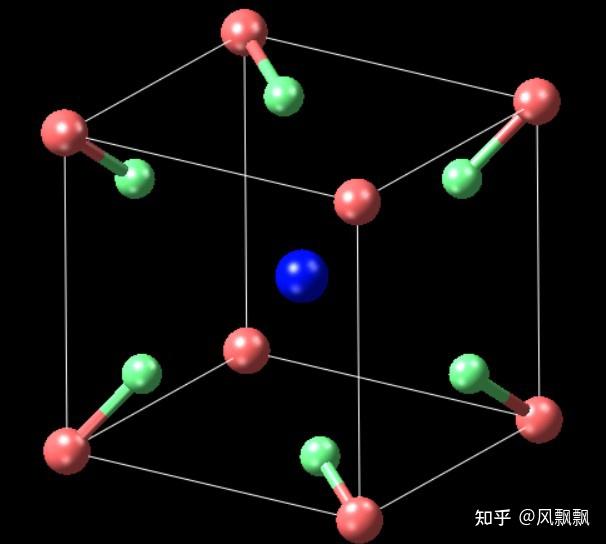
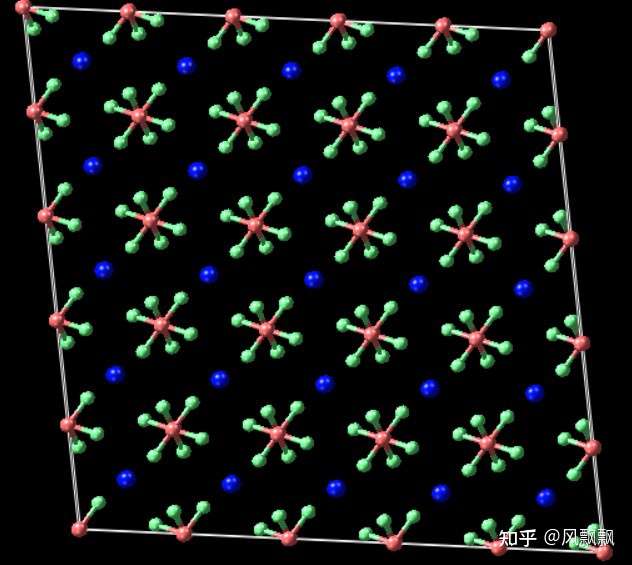
KPF6 crystal
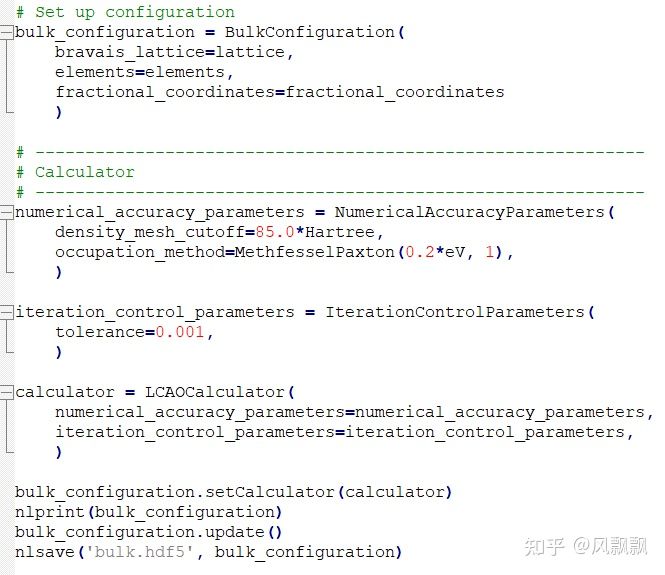

The script

The trajectory of KPF6
Simulation conditions:
1000 atoms(5x5x5);
time_step=1*femtoSecond,
reservoir_temperature=350*Kelvin,
reservoir_pressure=[1000, 1000, 1000, 1000, 1000, 1000]*bar,
thermostat_timescale=100*femtoSecond,
barostat_timescale=1000*femtoSecond,
initial_velocity=initial_velocity,
heating_rate=0*Kelvin/picoSecond,
compression_rate=0*bar/femtoSecond,
coupling_mask=[[True, True, True], [True, True, True], [True, True, True]],
chain_length=3,
1. Why does the simulation box eventually become so distorted? According to the phase diagram, KPF6 crystal may undergo phase transformation. Is it necessary to define the simulation box as a triclinic system?
2. The chemical bond of PF6 has broken, but it is obvious that the chemical bond of PF6 will not break at this temperature.
3. Are these results due to the inaccuracy of LCAO? However, the AIMD based on LCAO should predict the physical laws, even though the value may have some deviation.
I have been troubled by this problem for a long time and can't solve it with all kinds of methods. If someone can help me solve it, I will thank him very much.


